
Zaburzenie homeostazy lipidowej w deficycie lizosomalnej lipazy – patomechanizm, diagnostyka i leczenie
DOI:
https://doi.org/10.18388/pb.2021_389Abstrakt
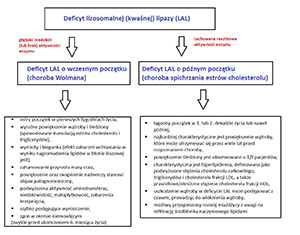
Lipaza lizosomalna katalizuje reakcję hydrolizy estrów cholesterolu i triglicerydów, której produktem są wolne kwasy tłuszczowe i wolny cholesterol, będące kluczowymi mediatorami wewnątrzkomórkowej homeostazy lipidowej. Deficyt LAL jest chorobą monogenową, spowodowaną obecnością patogennych wariantów molekularnych w genie LIPA, o dziedziczeniu autosomalnym recesywnym. W deficycie LAL dochodzi do kumulacji estrów cholesterolu i trójglicerydów w lizosomach, co wtórnie powoduje zwiększoną syntezę endogennego cholesterolu, apolipoproteiny B oraz lipoprotein o niskiej gęstości (VLDL). Diagnostyka deficytu kwaśnej lipazy jest łatwa, dzięki dostępnej obecnie metodzie badania aktywności enzymu w suchej kropli krwi. Analiza molekularna jest niezbędna celem weryfikacji diagnozy klinicznej i biochemicznej oraz analizy korelacji genotyp-fenotyp. Sebelipaza alfa jest rekombinowaną ludzką lizosomalną lipazą przeznaczoną do stosowania w enzymatycznej terapii zastępczej u pacjentów z niedoborem LAL.
Pobrania
Opublikowane
Numer
Dział
Licencja
Prawa autorskie (c) 2021 Postępy Biochemii

Utwór dostępny jest na licencji Creative Commons Uznanie autorstwa 4.0 Międzynarodowe.
Zawartość kwartalnika jest rozpowszechniana na licencji Creative Commons Uznanie autorstwa 4.0 Międzynarodowe (CC BY 4.0). Uznanie autorstwa — Utwór należy odpowiednio oznaczyć, podać link do licencji i wskazać jeśli zostały dokonane w nim zmiany . Możesz to zrobić w dowolny, rozsądny sposób, o ile nie sugeruje to udzielania przez licencjodawcę poparcia dla Ciebie lub sposobu, w jaki wykorzystujesz ten utwór.
Prawa autorskie do prac © pozostają przy autorach.
Prawa autorskie do czasopisma © posiada Polskie Towarzystwo Biochemiczne.

