
Disturbance of lipid homeostasis in lysosomal lipase deficiency – pathomechanism, diagnosis and treatment
DOI:
https://doi.org/10.18388/pb.2021_389Abstract
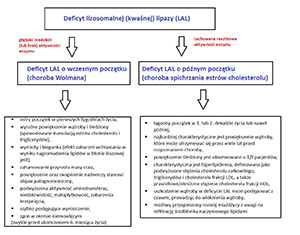
Lysosomal acid lipase (LAL) plays a key role in lipid metabolism through the hydrolysis of cholesteryl esters and triglycerides in lysosomes. LAL deficiency is a rare autosomal recessive lysosomal storage disease caused by deleterious mutations in the LIPA gene. In the case of LAL deficiency, cholesteryl esters and triglycerides accumulate within the lysosomes. The up-regulation of endogenous cholesterol production, increased synthesis of apolipoprotein B (ApoB) and increased production of very-low-density lipoprotein cholesterol (VLDL-C) is observed. The diagnosis is easy due to the currently available method of testing the enzyme activity in a dry blood spot. Molecular analysis is necessary to verify the clinical and biochemical diagnosis and to analyze the genotype-phenotype correlation. Sebelipase alfa is a recombinant human lysosomal lipase intended for use in enzyme replacement therapy in patients with LAL deficiency.
Published
Issue
Section
License
Copyright (c) 2021 Advances in Biochemistry

This work is licensed under a Creative Commons Attribution 4.0 International License.
All journal contents are distributed under the Creative Commons Attribution-ShareAlike 4.0 International (CC BY-SA 4.0) license. Everybody may use the content following terms: Attribution — You must give appropriate credit, provide a link to the license, and indicate if changes were made, ShareAlike — If you remix, transform, or build upon the material, you must distribute your contributions under the same license as the original. There are no additional restrictions — You may not apply legal terms or technological measures that legally restrict others from doing anything the license permits.
Copyright for all published papers © stays with the authors.
Copyright for the journal: © Polish Biochemical Society.
![]()

