Molecular mechanisms of Duchenne muscular dystrophy and new therapeutic strategies
DOI:
https://doi.org/10.18388/pb.2021_428Abstract
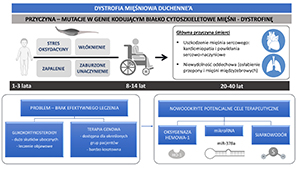
Duchenne muscular dystrophy (DMD) is an X-linked genetic disease affecting approximately 1 in 5,000 born boys. It is caused by mutations in the DMD gene encoding dystrophin, which protects muscle fibers upon contraction. Its absence leads to muscle weakening and premature death mostly due to cardio-respiratory failure. Many experimental therapies have been developed to restore functional dystrophin or counteract processes contributing to disease progression. Nonetheless, DMD remains an incurable disease, and glucocorticoids, exerting many side effects, still serve as the “gold standard” of treatment. Hence, there is a need to develop innovative therapeutic options that will at least alleviate the symptoms of DMD. Among them, targeting specific microRNAs (miRs), e.g. miR-378a, restoring normal angiogenesis and the use of cytoprotective factors such as heme oxygenase-1 (HO-1) or hydrogen sulfide (H2S) might be of special interest. In this review, we describe both the pathology of the disease and the aforementioned new therapeutic options in DMD.
Downloads
Published
Issue
Section
License
Copyright (c) 2022 Paulina Podkalicka, Małgorzata Myszka, Józef Dulak, Agnieszka Łoboda

This work is licensed under a Creative Commons Attribution 4.0 International License.
All journal contents are distributed under the Creative Commons Attribution-ShareAlike 4.0 International (CC BY-SA 4.0) license. Everybody may use the content following terms: Attribution — You must give appropriate credit, provide a link to the license, and indicate if changes were made, ShareAlike — If you remix, transform, or build upon the material, you must distribute your contributions under the same license as the original. There are no additional restrictions — You may not apply legal terms or technological measures that legally restrict others from doing anything the license permits.
Copyright for all published papers © stays with the authors.
Copyright for the journal: © Polish Biochemical Society.
![]()