
Molekularne mechanizmy dystrofii mięśniowej Duchenne’a i nowe możliwości terapeutyczne
DOI:
https://doi.org/10.18388/pb.2021_428Abstrakt
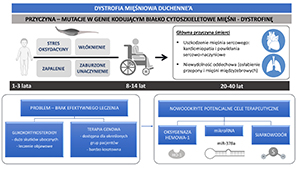
Dystrofia mięśniowa Duchenne’a (DMD) to choroba genetyczna sprzężona z chromosomem X, dotykająca w przybliżeniu 1 na 5000 urodzonych chłopców. Jest spowodowana mutacjami w genie DMD kodującym dystrofinę, która odpowiada za stabilność mechaniczną mięśni podczas skurczu. Jej brak prowadzi do postępującego osłabienia mięśni i przedwczesnej śmierci chorych w wyniku niewydolności sercowo-oddechowej. W ostatnich latach opracowano wiele eksperymentalnych terapii, których celem jest przywrócenie funkcjonalnej dystrofiny lub przeciwdziałanie procesom przyczyniającym się do postępu choroby, takim jak zapalenie czy zwłóknienie. Pomimo tego DMD wciąż pozostaje chorobą nieuleczalną, a glikokortykoidy, wykazujące wiele działań niepożądanych, nadal stanowią „złoty standard” leczenia. Aktualne jest zatem opracowywanie innowacyjnych możliwości terapeutycznych, które przynajmniej złagodzą objawy DMD. Wśród nich na uwagę zasługuje celowanie w określone mikroRNA (miR), np. miR-378a, przywrócenie prawidłowej angiogenezy oraz wykorzystanie cytoprotekcyjnych czynników takich jak oksygenaza hemowa-1 (HO-1) czy siarkowodór (H2S). W niniejszej pracy omówiono zarówno patologię choroby jak i wspomniane, nowe możliwości terapeutyczne w DMD.
Opublikowane
Numer
Dział
Licencja
Prawa autorskie (c) 2022 Paulina Podkalicka, Małgorzata Myszka, Józef Dulak, Agnieszka Łoboda

Utwór dostępny jest na licencji Creative Commons Uznanie autorstwa 4.0 Międzynarodowe.
Zawartość kwartalnika jest rozpowszechniana na licencji Creative Commons Uznanie autorstwa 4.0 Międzynarodowe (CC BY 4.0). Uznanie autorstwa — Utwór należy odpowiednio oznaczyć, podać link do licencji i wskazać jeśli zostały dokonane w nim zmiany . Możesz to zrobić w dowolny, rozsądny sposób, o ile nie sugeruje to udzielania przez licencjodawcę poparcia dla Ciebie lub sposobu, w jaki wykorzystujesz ten utwór.
Prawa autorskie do prac © pozostają przy autorach.
Prawa autorskie do czasopisma © posiada Polskie Towarzystwo Biochemiczne.

