
Symulacje chemicznych układów kwantowych z wykorzystaniem komputerów kwantowych – przegląd algorytmów i ich eksperymentalna weryfikacja
DOI:
https://doi.org/10.18388/pb.2021_536Abstrakt
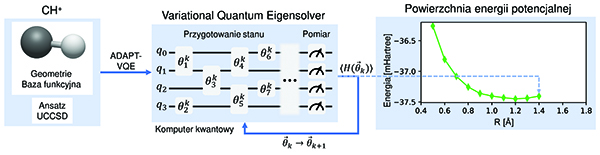
Symulacje komputerowe wykorzystujące coraz większe moce obliczeniowe i techniki uczenia maszynowego pozwalają obecnie na zaawansowane modelowanie molekularne, symulacje dynamiki molekularnej oraz badania interakcji międzycząsteczkowych. Ze względu jednak na złożoność systemów biologicznych i procesów chemicznych na poziomie molekularnym ich dokładne odwzorowanie z wykorzystaniem klasycznych modeli i technik komputerowych napotyka od wielu lat szereg istotnych ograniczeń. Nowym i obiecującym kierunkiem rozwoju nauk obliczeniowych oraz ich potencjalnych zastosowań w biochemii są komputery kwantowe oraz ich integracja z klasycznymi, wysokowydajnymi systemami superkomputerowymi. Artykuł jest odpowiedzią na rosnące zainteresowanie wykorzystanie dostępnych komputerów kwantowych w przykładowych zastosowaniach aplikacyjnych. W artykule staramy się przybliżyć podstawowe zagadnienia związane z opracowaniem algorytmów i symulacji kwantowych związanych z zagadnieniami na styku chemii kwantowej oraz biochemii. Ponadto artykuł przybliża podstawowe zasady tworzenia symulacji wykorzystujących obecny stan zaawansowania i rozwoju komputerów kwantowych w erze Noisy Intermediate-Scale Quantum (NISQ). W artykule przedstawiono również wyniki eksperymentalne algorytmu klasyczno-kwantowego Variational Quantum Eigensolver (VQE) dla przykładowych cząsteczek H2 i CH+. Pomimo wielu niedoskonałości obecnie dostępnych komputerów kwantowych, analizowany algorytm VQE okazał się skuteczny w przybliżaniu stanu podstawowego cząsteczek, wykorzystując minimalną bazę funkcyjną.
Pobrania
Opublikowane
Licencja
Prawa autorskie (c) 2024 Konrad Wojciechowski, Krzysztof Kurowski, Cezary Mazurek

Utwór dostępny jest na licencji Creative Commons Uznanie autorstwa 4.0 Międzynarodowe.
Zawartość kwartalnika jest rozpowszechniana na licencji Creative Commons Uznanie autorstwa 4.0 Międzynarodowe (CC BY 4.0). Uznanie autorstwa — Utwór należy odpowiednio oznaczyć, podać link do licencji i wskazać jeśli zostały dokonane w nim zmiany . Możesz to zrobić w dowolny, rozsądny sposób, o ile nie sugeruje to udzielania przez licencjodawcę poparcia dla Ciebie lub sposobu, w jaki wykorzystujesz ten utwór.
Prawa autorskie do prac © pozostają przy autorach.
Prawa autorskie do czasopisma © posiada Polskie Towarzystwo Biochemiczne.

